
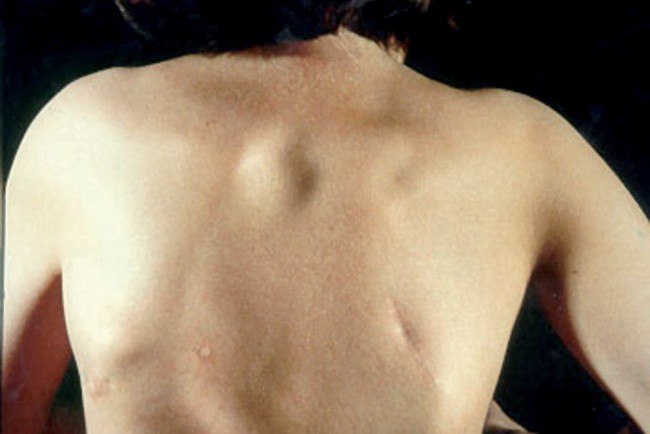
Une maladie quasi-impossible à diagnostiquer
Du fait de sa rareté et du manque de connaissances vis-à-vis de ses mécanismes, la maladie de l’homme de pierre est une affection extrêmement difficile à dépister, et qui apparaît dès l’enfance. Mise à part une malformation du gros orteil, courbé et de petite taille, rien n’indique que l’enfant est atteint de fibrodysplasie ossifiante progressive. Ce n’est qu’entre deux et cinq ans que les premières manifestations sont observées. Bien entendu, face à l’impossibilité de diagnostiquer efficacement et de comprendre tous les rouages de la maladie, aucun traitement n’a encore été élaboré. D’ailleurs, dans l’état actuel des connaissances liées à cette affection, toute intervention chirurgicale aggrave la condition du patient plus qu’elle ne l’améliore ; si l’on retire une partie de la formation osseuse, celle-ci est comblée par l’apparition d’une nouvelle plaque, encore plus importante et plus solide, exactement comme lorsqu’un os fracturé se reforme de lui-même. Dans les cas les plus graves, une simple vaccination ou une prise de sang entraînent de graves complications.
Les causes de la maladie
La maladie de l’homme de pierre aurait pour origine une anomalie génétique complexe. En effet, le gène qui code le récepteur ACVR1 (le récepteur étant une protéine située à la surface de la cellule et permettant à celle-ci d’échanger des informations) subirait une mutation. Or, le récepteur ACVR1 serait la protéine engagée dans la formation et la structure des os (ou morphogenèse osseuse). En mutant, le gène codant le récepteur ACVR1 entraîne donc le dysfonctionnement de ce dernier.


